Atomwise scientists are improving predictive models for ADMET profiling by including a pre-training step that builds on diverse publicly available datasets
For any candidate drug, pharma scientists need to answer an important question: how does the body respond to the compound? Once scientists identify a molecule that binds to their protein of interest and seems a likely drug candidate, they have to assess its absorption, distribution, metabolism, excretion, and toxicity (ADMET) profile. These tests let scientists understand the solubility of the compound in the body, how slowly it clears, its distribution in the body, and its toxicity to healthy cells.
Synthesizing drug compounds and then testing them in in vitro and in vivo assays is both time-consuming and expensive. To keep costs down, drug discovery scientists can use machine learning approaches to predict the ADMET profiles of the compounds in which they are interested before investing in real-world experiments. This way, scientists only test compounds that show the greatest promise, filtering out the ones least likely to have favorable ADMET properties.
One of the challenges with this task is that the ADMET datasets available for training predictive models are often quite small. To resolve this, Atomwise scientists are working on a way to build generalizable predictive models based on graph neural networks (GNNs) using available ADMET data. Their approach involves a pre-training step that uses a large pool of publicly available compounds to provide baseline learning for the GNN models. The models are pre-trained to model important physicochemical properties of the compound and the presence of different functional groups. This is followed by a second step where the models are fine-tuned using the much smaller ADMET datasets to estimate properties of interest for future drug-like compounds.
 Developing this kind of capability is a logical next step for Atomwise because it extends the company’s computational capability into new territory, says Hossam Ashtawy, a machine learning scientist at Atomwise. “Atomwise started with technologies that allow you to identify bioactive compounds that bind to the protein of interest and now we have expanded to ADMET prediction as well.”
Developing this kind of capability is a logical next step for Atomwise because it extends the company’s computational capability into new territory, says Hossam Ashtawy, a machine learning scientist at Atomwise. “Atomwise started with technologies that allow you to identify bioactive compounds that bind to the protein of interest and now we have expanded to ADMET prediction as well.”
From the raw descriptors of molecules such as atom types and the bonds between them, neural networks can learn higher-level molecular representations of drug compounds tailored for the tasks of interest such as assessing ADMET. These automatic representations tend to provide better predictive accuracy than their manually-engineered counterparts that have been traditionally used in drug discovery applications. But how well these networks can learn these representations depends on their architecture and the quality of the data used for training. Larger neural networks tend to generate better representations and hence predictive models, but their effectiveness depends on the size of the dataset, diversity, and how much noise it contains.
Ideally, the dataset should include a minimum of tens of thousands of compounds, Ashtawy says. It is also important to have a diversity of compounds in the training dataset. “If all of those compounds are centered around a few scaffolds in the chemical space, then the resulting model may not generalize well to an extended chemical space,” he adds.
Access to better ADMET data should result in more accurate representations of the compounds by the models during the training process. But there is a scarcity of high-quality datasets in public databases which makes training directly on ADMET data difficult. The parameters of a neural network are typically initialized to random values prior to training and as a result, the initial representations are not useful. “When you train, the representations become better, but if your training data is not big enough, the neural networks may not converge to good representations,” Ashtawy says.
Atomwise’s solution is to pre-train the models first using a cadre of compounds that cover a large chemical space. This provides a baseline on which to build. Our scientists used several public and private libraries for pre-training, including data from resources such as the ChEMBL database and companies such as Enamine and Mcule. They sampled a total of 20 million diverse compounds.
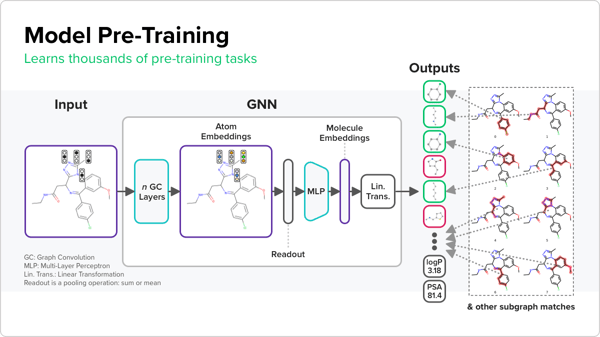
From this first step, the GNNs can generate representations that encode for the presence or absence of thousands of diverse sub-structures as well as various important physicochemical properties of the molecules. Then, when the team trains the models using the much smaller ADMET dataset, the hope is that the final representations of the molecule will more accurately represent its ADMET profile. A final ADMET predictive model is produced by modeling the existing experimental data using the representations as input.
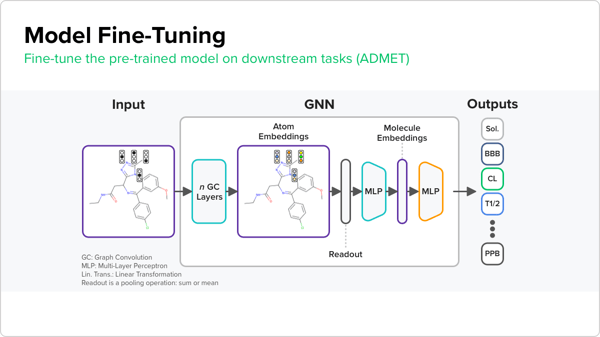
It is a simpler and more effective approach than some other pre-training techniques described in the scientific literature, Ashtawy says. Preliminary results from using the pre-trained models are promising: Atomwise scientists observed a boost in performance with the pre-trained models for different endpoints compared to models that were not pre-trained. Ultimately, these models could be used to test different kinds of drugs in various disease areas. Atomwise has already begun applying the trained models to specific drug targets as part of an internal portfolio.
“Any company that plans to test drugs on humans and apply them to human health will have to show that they are safe, efficacious, and have good ADMET properties,” Ashtawy says. “Using predictive models that can accurately estimate these parameters will lead to a significant reduction in development time and cost of promising drugs in many disease areas.”
Learn More
Dr. Hossam Ashtawy was selected to present this topic at the American Chemical Society Fall 2021 Virtual Meeting & Expo. Take a deeper dive by viewing his presentation deck: Pretraining Graph Neural Networks on Ultra Large Chemical Libraries to Learn Generalizable ADMET Predictors
About Atomwise
Atomwise is a technology-enabled pharmaceutical company leveraging the power of AI to revolutionize small molecule drug discovery. The Atomwise team invented the use of deep learning for structure-based drug design; this technology underpins Atomwise’s best-in-class AI discovery engine, which is differentiated by its ability to find and optimize novel chemical matter.
Atomwise has extensively validated its discovery engine, delivering success in over 185 projects to-date including a wide-variety of protein types and numerous “hard-to-drug” targets. Atomwise is building a wholly-owned pipeline of small-molecule drug candidates, with three programs in lead-optimization and over 30 programs in discovery.
The company has raised over $174 million from leading venture capital firms to advance its mission to make better medicines, faster.
Learn more at atomwise.com, or connect on Twitter and LinkedIn.
