Atomwise scientists are adapting machine learning models to make predictions about a class of drug compounds that has been challenging to study.
Drug compounds that act as covalent inhibitors can modulate protein function with more potency, selectivity, and longevity than non-covalent ones. However, pharmaceutical companies have historically deprioritized these compounds in their drug discovery programs because of concerns about their toxicity and off-target effects.
“These covalent interactions release a lot of energy, so they are very strong. One unintended aspect of these strong interactions is that they become less specific so they hit every protein,” says Srimukh Prasad, a cheminformatics scientist at Atomwise. Scientists also worried these compounds would be a risky investment because they did not fully understand their mechanisms of action.
However, there is a resurgent interest in these compounds. In the last decade, the U.S. Food and Drug Administration has approved 14 covalent drugs, including Amgen’s Lumakras, a covalent inhibitor used to treat adults with some forms of non-small cell lung cancer.
At Atomwise, the growing interest in covalent inhibitors presents an exciting opportunity for scientists to extend the capabilities of our current deep learning-based virtual high-throughput screening pipelines. These pipelines routinely screen billions of compounds against protein targets of interest. But the underlying assumption of the models is that the interaction between the protein target and the compound is purely non-covalent in nature. Working on covalent inhibitors posed a different set of challenges that our models would need to overcome to screen them.
The first challenge was curating the data our machine learning models require. Our scientists have trained the models to predict non-covalent inhibitors using only data on non-covalent binding. To make the models aware of covalent inhibition, they needed to train the models to use both non-covalent and covalent inhibition data. However, there is currently not enough information about the mechanisms of the compound-protein interactions for the models to classify bonds as covalent. The models need this information to generate accurate docking conformations.
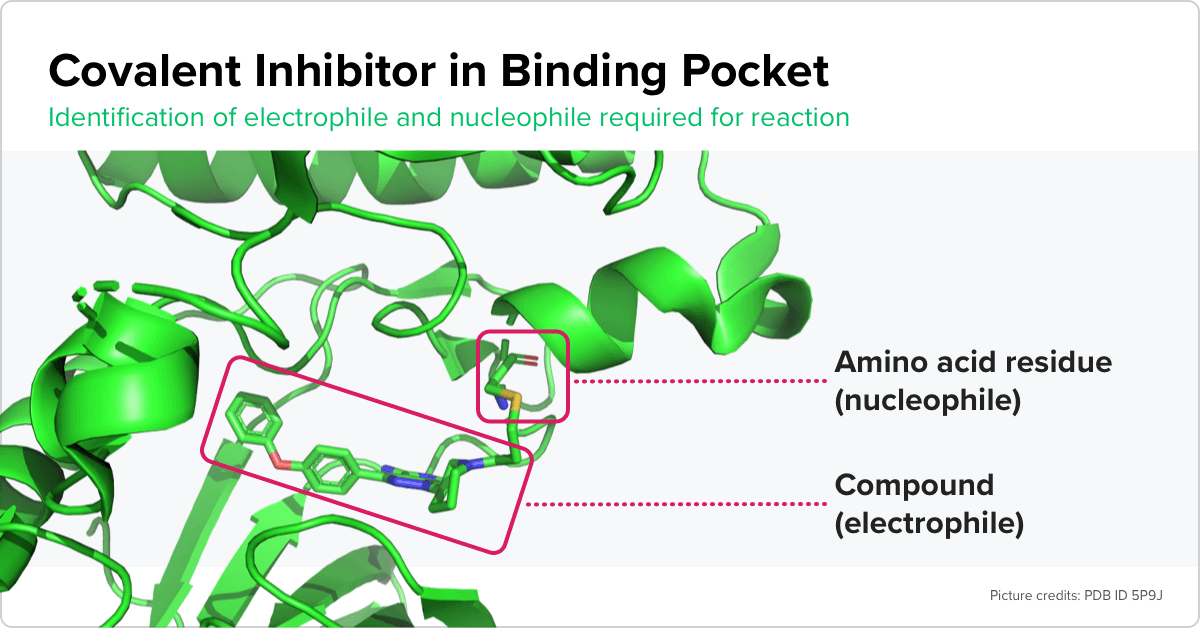
Atomwise scientists employed a unique approach to generate this data. Covalent inhibitors have specific electrophilic functional groups called warheads that play an important role in their interactions with the target proteins. Target proteins that are amenable to covalent interactions must have specific nucleophilic amino acid residues in their binding pocket. Prasad first set out to automatically label interactions between hundreds of thousands of compounds and proteins that met these criteria as covalent.
The second challenge was figuring out how to modify Atomwise’s AtomNet® deep learning models to identify covalent inhibitors without compromising their ability to predict interactions with non-covalent compounds. The AtomNet® models account for the three-dimensional structures of the protein, the compound, and the potential pose of the compound when it is bound to the protein pocket. However, the models lacked the capability to incorporate the unique characteristics of covalent inhibitors.
To capture these features, Atomwise scientists added a constrained docking algorithm to CUina, their GPU-accelerated software for modeling protein-compound interactions. The constraint mimics the covalent bonds and generates docking poses for covalent compounds.
The algorithm incorporates scientists’ understanding of covalent molecules and mechanisms of interaction, adding information on warhead-protein residue interactions and the distance between these chemical groups. Using this information, the model can make predictions about covalent inhibitors in a high-throughput manner.

“While the covalent inhibitor forms a covalent bond with the protein target, it also interacts with the target non-covalently,” Prasad says. “Our CUina docking tool takes both covalent and non-covalent interactions into account.” The final poses generated by the CUina docking tool are used to train and validate a deep-learning virtual high-throughput screening model for identifying covalent inhibitors.
It's early days for this project. There is still work to do to train and test Atomwise’s covalent models as well as to ensure that the adaptations do not hinder their ability to predict proteins interacting non-covalently with molecules. But the first results have been promising. The quality of compound-protein docking poses for covalent compounds generated using the Atomwise models is comparable to published covalent docking protocols.
“This gives us the ability to develop promising compounds for classes of proteins that are difficult to target non-covalently,” Prasad says. “We hope this will lead to better therapeutics and improved patient outcomes.”
Learn MoreDr. Srimukh Prasad was selected to present on this topic at the American Chemical Society Fall 2021 Virtual Meeting & Expo. Take a deeper dive by viewing his presentation deck: https://blog.atomwise.com/a-virtual-high-throughput-screening-pipeline-for-covalent-inhibitors
About Atomwise
Atomwise is a technology-enabled pharmaceutical company leveraging the power of AI to revolutionize small molecule drug discovery. The Atomwise team invented the use of deep learning for structure-based drug design; this technology underpins Atomwise’s best-in-class AI discovery engine, which is differentiated by its ability to find and optimize novel chemical matter.
Atomwise has extensively validated its discovery engine, delivering success in over 185 projects to-date including a wide-variety of protein types and numerous “hard-to-drug” targets. Atomwise is building a wholly-owned pipeline of small-molecule drug candidates, with three programs in lead-optimization and over 30 programs in discovery.
The company has raised over $174 million from leading venture capital firms to advance its mission to make better medicines, faster.
Learn more at atomwise.com, or connect on Twitter and LinkedIn.
