Our scientists developed AtomNet® PoseRanker to improve our technology’s ability to rank poses for protein-ligand interactions with potential drug compounds
Identifying the highest-quality protein-ligand poses generated from structure-based virtual high-throughput screening (vHTS) approaches is a major challenge for drug discovery. These methods typically use a physics-based scoring function to generate ranked lists of plausible poses, which are models of physical interactions between small molecules and protein targets. They are often effective at suggesting good poses, but the best ones are typically not at the top of the list. Also, the suggested poses, generated from crystal structures in a single conformation, may not be the right match for proteins in vivo.
To address these challenges, our scientists developed AtomNet® PoseRanker (ANPR), a new tool based on our AtomNet® technology. ANPR uses deep learning-based methods to identify the best poses for interactions between proteins and small molecules of interest. It significantly outperforms existing physics-based docking methods. Earlier this year, our team published a paper describing ANPR in the Journal of Chemical information and Modeling.
The area of protein dynamics is of great interest to Kate Stafford, Atomwise’s director of structural cheminformatics. During her doctoral research at Columbia University, she worked on molecular dynamics simulations of proteins with flexible regions that determine substrate binding. Later, during her postdoctoral fellowship at University of California, San Francisco, she used molecular docking to identify small molecules likely to bind to protein targets. These kinds of studies commonly rely on tools like Autodock Vina and an open-source implementation, Smina, which use a simplified force field to dock small molecules to proteins of interest.
The latest results from published benchmarks show that these tools are great at generating possible models of physical interactions between small molecules and protein targets — but they are not very good at ranking the best ones at the top of the list. Our scientists had the same experience when they used the tools. “We get a ranked list of potential poses and models of physical interactions and somewhere in that ensemble there was typically a good pose but it was not ranked at the top,” she says.
Our scientists also noticed problems with using a single static crystal protein structure in docking experiments. Sometimes, they were unable to find a good pose at all because the protein conformations they were trying to dock were not quite the right fit. “We noticed that really good results from molecular docking required more than a single protein conformation,” Stafford says.
Unlike traditional, structure-based virtual high-throughput screening approaches that rely on a single crystal structure, ANPR uses ensembles of conformations. These ensembles include a reference crystal structure and several similar low-energy structures. “We set out to improve our enrichment over physics-based docking methods,” Stafford says. “In this instance, enrichment means that it is more likely that a good pose shows up higher in the ranked list of poses, across the ensemble.”
Atomwise scientists used reference data from PDBbind, a curated database of protein crystal structures bound to small molecules and their measured activities to train ANPR. To create the training data, the team used docked poses for the proteins in the reference. The team also sampled additional conformations for each protein in the training dataset. “We generated lots of poses in our ensembles, some that were inaccurate, to make sure that we sampled some good ones,” Stafford says.
The model is trained to identify poses that are within a cutoff based on a gold standard. Once trained, the model reports a probability that each pose is within the cutoff.
Strikingly, by exposing ANPR to ensembles of protein conformations with docked compounds, the model seemed to learn how proteins move to accommodate ligand binding. “What we found interesting and surprising is that we were able to improve the prediction of activity of compounds if we only did inference on a single structure,” Stafford says. “This means that the model learned something about protein flexibility from training on additional conformations. But when you are actually using the model, you can do the inference from a single structure.”
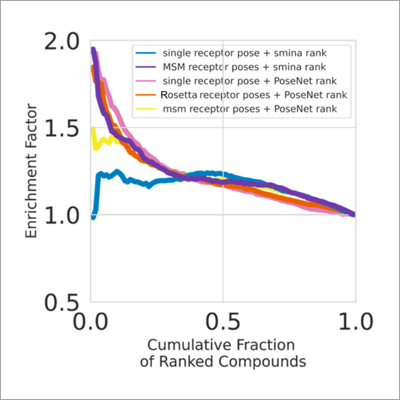
ANPR pose ranking enriched known Abl inhibitors compared to smina. Enrichment persisted even for single receptor conformations. It suggests that ANPR learned to infer and account for protein receptor flexibility when recognizing high quality poses.
Reference: https://pubs.acs.org/doi/10.1021/acs.jcim.1c01250#
One of our scientists’ goals for this project was to make better predictions of protein-small molecule activity by improving the quality of physics-based interaction models.

“We have made our set of sampled conformations publicly available so that people who are interested in following up on this observation about the benefits of conformational sampling have the data already prepared for them,” Stafford says. The data is available here.
Our scientists have also published a follow-up study that shows how ANPR improves virtual screening when it is used as part of the training process. “We show that when you provide a better model of the physics, you get better predictions of activity,” Stafford adds. “ANPR makes the virtual screening process much more sensitive to the underlying physics.”
Learn MorePublication: Kate A. Stafford, Brandon M. Anderson, Jon Sorenson, and Henry van den Bedem, AtomNet PoseRanker: Enriching Ligand Pose Quality for Dynamic Proteins in Virtual High-Throughput Screens, J. Chem. Inf. Model. 2022, 62, 5, 1178–1189 Publication Date:March 2, 2022
https://pubs.acs.org/doi/10.1021/acs.jcim.1c01250
About Atomwise
Atomwise is a technology-enabled pharmaceutical company leveraging the power of AI to revolutionize small molecule drug discovery. The Atomwise team invented the use of deep learning for structure-based drug design; this technology underpins Atomwise’s best-in-class AI discovery engine, which is differentiated by its ability to find and optimize novel chemical matter.
Atomwise has extensively validated its discovery engine, delivering success in over 185 projects to-date including a wide-variety of protein types and numerous “hard-to-drug” targets. Atomwise is building a wholly-owned pipeline of small-molecule drug candidates, with three programs in lead-optimization and over 30 programs in discovery.
The company has raised over $174 million from leading venture capital firms to advance its mission to make better medicines, faster.
Learn more at atomwise.com, or connect on Twitter and LinkedIn.
