With the programmers on the Atomwise team, we have a steady stream of new ideas for how to improve the AtomNet® platform we created to virtually screen protein targets against billions of compounds to find promising drug candidates.
 At a recent meeting of the American Chemical Society, Atomwise team members presented results from some of our latest efforts to boost efficiency and enhance predictions. Brandon Anderson, a physicist by training and head of the ml.research group at Atomwise, gave a talk about his work to improve pose classification — how AtomNet® technology determines whether the target and compound fit together — using SO(3)-equivariant neural networks.
At a recent meeting of the American Chemical Society, Atomwise team members presented results from some of our latest efforts to boost efficiency and enhance predictions. Brandon Anderson, a physicist by training and head of the ml.research group at Atomwise, gave a talk about his work to improve pose classification — how AtomNet® technology determines whether the target and compound fit together — using SO(3)-equivariant neural networks.
Anderson came from the world of physics, and then made the switch to physics-inspired neural networks, in which he focused on how to incorporate physical symmetries into these networks. “If you have a molecule and you want to learn some property of it, such as the distance between atoms, then you want to make sure if you move the molecule around in space in a virtual environment that you get the correct result,” he says.
Physics appealed to him early on because of the fascinating challenge it presented. He took a quantum mechanics class in college. “I found it mind-bending that abstract math could model unintuitive phenomena in the real world,” he recalls. “That’s when I knew I wanted to work on mathematically related physics.”
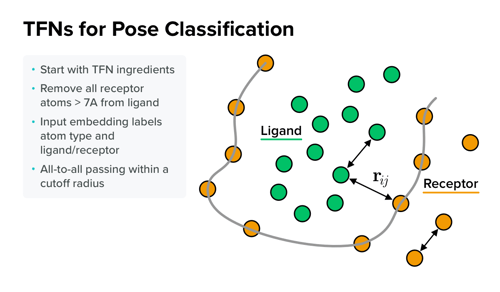
When Anderson joined Atomwise, he brought significant expertise in modeling, which is an important element in the AtomNet® platform’s ability to correctly predict whether targets and compounds would fit together. “Within the pipeline, one of the steps is to dock the ligand into the protein and determine if that docking is good or not,” he says. For optimal results, the virtual process must represent what would happen in the real world, perfectly mirroring how atoms would interact between the ligand and protein. “Local changes in geometry are very important for this,” he adds.
Anderson chose Tensor Field Networks (TFNs) to enable rotationally-equivariant features for both the ligand and receptors. “We want to design a neural network where the structure of the network closely resembles physical reality,” he says. “So each atom at each layer of the network has a representation of its local environment. These representations encode geometric information about what atoms are near it and how they relate to each other in space.”
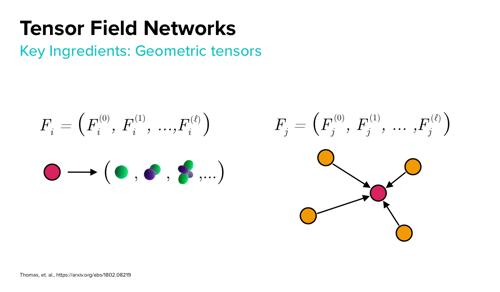
His approach then combines the ligand and receptor features into a single interaction network. In his presentation at ACS, he compared results of this technique to metrics from open-source screening benchmarks and demonstrated that it performed well. “It seemed to outperform our best pose prediction by nontrivial margin,” he says.
 One built-in advantage to Anderson’s method is that it is computationally efficient because the network can learn about and model local geometry with fewer parameters. Next, he plans to explore TFNs as a means to improve binding affinity prediction, hoping that will benefit from geometric representations as well.
One built-in advantage to Anderson’s method is that it is computationally efficient because the network can learn about and model local geometry with fewer parameters. Next, he plans to explore TFNs as a means to improve binding affinity prediction, hoping that will benefit from geometric representations as well.
Learn More
Brandon Anderson, PhD, was selected to present on this topic at the American Chemical Society Spring 2021 Virtual Meeting & Expo. Take a deeper dive by viewing his presentation deck: Protein-Ligand Binding Predictions with SO(3)-Equivariant Neural Networks
About Atomwise
Atomwise is a preclinical pharma company revolutionizing how drugs are discovered with AI. We invented the use of deep learning for structure-based drug discovery, today developing a pipeline of small-molecule drug candidates advancing into preclinical studies. Our AtomNet® technology has been used to unlock more undruggable targets than any other AI drug discovery platform. We are tackling over 600 unique disease targets with more than 250 partners around the world, including leading pharmaceutical, agrochemical, and emerging biotechnology companies. Atomwise has raised over $174 million from leading venture capital firms to advance our mission to make better medicines, faster.
