If you think of virtual drug screening as a video game, with players lining up models of compounds and protein targets to see if they fit together, then binding affinity might be how the game is scored. In drug discovery, we look for strong binding affinity as an indicator that the compound will bind to the target and cause the desired biological effect.
In our AtomNet® technology virtual screening tool — which is, by the way, even cooler than a video game — calculating binding affinity is a very important part of how the AI predicts which compounds are the best candidates to bind to a target. Our computational team is constantly working to improve the way our algorithms model this interaction.
 At a recent ACS meeting, Atomwise principal scientist Michael Mysinger gave a talk about the latest improvements we’ve achieved on this front. These efforts are largely built on a performance comparison of voxel-based and point-based convolutional neural network models for binding affinity, something known in the field as pKi modeling.
At a recent ACS meeting, Atomwise principal scientist Michael Mysinger gave a talk about the latest improvements we’ve achieved on this front. These efforts are largely built on a performance comparison of voxel-based and point-based convolutional neural network models for binding affinity, something known in the field as pKi modeling.
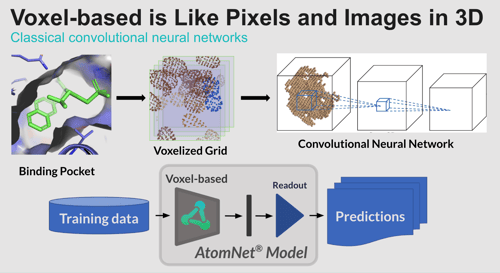
The AI community has largely been moving from voxel-based to point-based networks, and our results support that trend. In the former, the model sees everything on a voxel grid. A voxel-based network requires a significant amount of data augmentation where the protein and ligand have to be rotated to get different multiple viewpoints of the binding interaction. By contrast, point networks think about things in terms of atoms and the distances between them; rotation does not cause a problem for this type of model. We’ve found this approach to be a more natural fit for representing molecules.
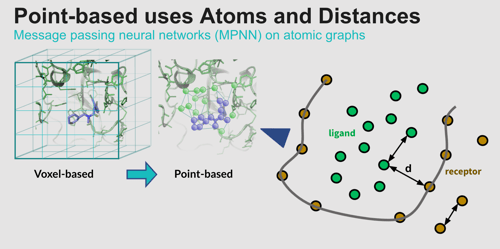
Point networks learn faster than voxel networks, which turns out to be both an advantage and a disadvantage. For the AtomNet® model, which has to screen billions of compounds, speed is very important. But point-based networks can learn so fast that they actually overfit the data. Our team addresses this by randomly dropping out up to 20% of the edges to make the model more flexible and better focused on the elements that matter most.
In our team’s comparison, point-based networks outperformed voxel-based networks for modeling binding affinity, in part because of the flexibility we build into the point networks. Mysinger notes that the AtomNet® platform’s task of modeling any protein or any ligand is “unique in the field.” Typically, virtual screening tools are purpose-built for one type of protein or a select number of ligands. “In our case, it’s one model that’s meant to predict all targets and all ligands,” he adds.
So how exactly does one test binding affinity predictions? Mysinger’s background in putting together benchmarking sets was instrumental. He designed a compound series benchmark, which involved identifying closely related compounds with known binding affinities. Our final set included about 7,000 series, each with anywhere from 10 to 600 ligands. Additionally, we judge the AI on how well it predicts protein targets distinct from those it has seen, specifically those with less than 70% sequence identity.

Because we know the binding affinities of all those compounds, we can rank-order them based on real-world data and then see how our network predictions compare. That’s how we determined that point networks perform better than voxel networks. It also demonstrates the broad applicability of our AtomNet® technology across huge families of compounds. “It’s a tough benchmark because the network has to generalize to a target it hasn’t seen before, but that’s important for us because we often tackle novel targets,” Mysinger says. “The point network performs well with this.”
Learn More
Michael Mysinger, PhD, was selected to present on this topic at the American Chemical Society Spring 2021 Virtual Meeting & Expo. Take a deeper dive by viewing his presentation deck - Comparing Voxel-Based and Graph-Based Convolutional Neural Network Models to Quantitatively Predict Binding Affinity Globally Across Structures and Ligands
About Atomwise
Atomwise is a preclinical pharma company revolutionizing how drugs are discovered with AI. We invented the use of deep learning for structure-based drug discovery, today developing a pipeline of small-molecule drug candidates advancing into preclinical studies. Our AtomNet® technology has been used to unlock more undruggable targets than any other AI drug discovery platform. We are tackling over 600 unique disease targets with more than 250 partners around the world, including leading pharmaceutical, agrochemical, and emerging biotechnology companies. Atomwise has raised over $174 million from leading venture capital firms to advance our mission to make better medicines, faster.
