In November, Atomwise published the first scientific paper about our technologies – AtomNet® platform: A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-based Drug Discovery. This blog post is an introduction to that work for a general audience. It assumes no prior scientific knowledge, and is greatly simplified for teaching purposes. Readers with scientific expertise should read the full paper.
Atomwise is known for using artificial intelligence to help discover new medicines. Historically, we have not revealed how we do that. Recently, we published the first details about one of our key technologies, a system we call AtomNet® model. AtomNet® technology marks a milestone for machine learning in the life sciences because it is the first deep convolutional neural network for structure-based, “rational” drug design. In this article, we want to introduce how AtomNet® technology works, share the thinking behind its creation, and explain why it is an important new drug discovery technology.
Drug Discovery is Expensive
Discovering a new medicine costs more and takes longer than at any point in history. In recent years, the average price-tag for getting a new drug to market has risen to about $2.5 billion, with an estimated delivery date of 10-15 years.
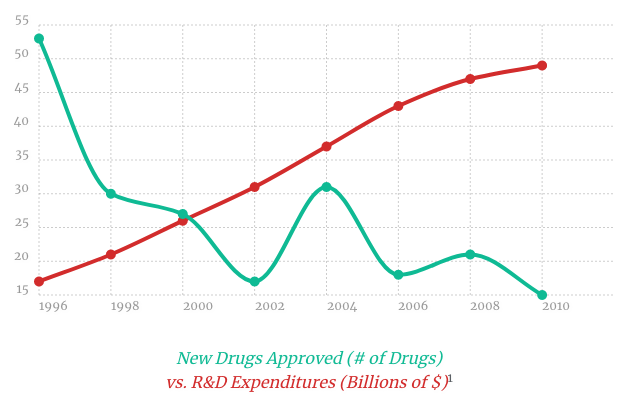
This trend is partly attributable to the high attrition rate of candidate molecules in the drug discovery process. For every molecule which becomes a drug, millions might be physically tested and discarded as unsuitable. (A dramatization would be to imagine if aerospace companies built and crashed thousands of planes for each design that stayed in the air.)
Meanwhile, urgent global health challenges, such as increasing antibiotic resistance and emerging pandemic viruses, only underscore that researchers need to shorten the time-to-discovery and explore more diverse chemical space to find new treatments. The same could be said for hard problems like Alzheimer’s disease that have challenged researchers for decades.
Drug Design vs. Drug Discovery
A logical response to the high failure rate of potential medicines would be to make smarter decisions about which ones we develop and test. If we understand the biological goal, perhaps molecules could be tailor-made for the task. This key concept is called Rational Drug Design.
In the life sciences, when a protein is known to play an important role in a disease, we call it a “target”. Examples of drug targets include proteins that cause inflammation, proteins that help tumors grow, or proteins that viruses use to infect human cells. In drug research, our goal is to create molecules that strongly interact with these targets, reducing (or enhancing) their effect. These molecules are called “ligands” to that target.
“In the most basic sense, drug design involves the design of molecules that are complementary in shape and charge to the biomolecular target with which they interact and therefore will bind to it.” A good metaphor is how keys fit into locks – there are billions of possible keys, but only a few open each specific lock.

Key opening a lock. 3
Ligand binding to a protein. 4
Keys vs. Locks
The “key and lock” model gives us a way to categorize the techniques that AtomNet® technology is inspired by. Lock-oriented techniques, which are called “structure-based” algorithms, look to the composition of the target protein to guide their predictions. This approach is appealing because, in principle, it works for totally novel targets. Accordingly, a variety of software packages have been introduced, such as Dock, AutoDock, and Glide. The main limitation of these technologies is their accuracy. In general, these methods have a high rate of false-positives (saying a molecule is a good candidate when it is not). Many researchers remain skeptical of their usefulness.
In contrast, some teams have looked to key-oriented techniques, which are called “ligand-based” algorithms. These technologies consider many examples of ligands that are already known to bind to a target, and ideally try to predict even better ligands from that data. Examples include ROCS and LINGOs. The main limitation of these technologies is that they require researchers to have already discovered at least some ligands for a target. However, for new, challenging, and unsolved drug discovery targets, there may be very few known ligands. In this case, these methods are expected to produce poor predictions.
An ideal solution then would be a structure-based rational drug design system that is also highly accurate. Such a system could predict candidate molecules for new and challenging drug discovery targets, and have a reasonable chance of those predictions proving correct – giving researchers what they need from a virtual drug discovery method. We think AtomNet® technology is a big step in that direction.
AtomNet® technology teaches itself college chemistry
AtomNet® technology is the first drug discovery algorithm to use a deep convolutional neural network. This type of network came to prominence only a few years ago and has a unique property: it excels at understanding complex concepts as a combination of smaller and smaller pieces of information. This property is a key reason why convolutional networks have produced the world’s best results for image classification, speech recognition, and other longstanding problems. For example, a convolutional model can learn to recognize faces by first learning a set of basic features in an image, such as edges. Then, the model can learn to identify parts such as noses, ears, and eyes by combining the edges. Finally, the model can learn to recognize faces by combining those parts.
Similarly, AtomNet® technology might learn that proteins and ligands are made up of a variety of specialized chemical structures. This would be an exciting result because it would suggest that AtomNet® model was learning fundamental concepts in organic chemistry. Intriguingly, this is what AtomNet® platform does. When we examine different neurons on the network we see something new: AtomNet® platform has learned to recognize essential chemical groups like hydrogen bonding, aromaticity, and single-bonded carbons.
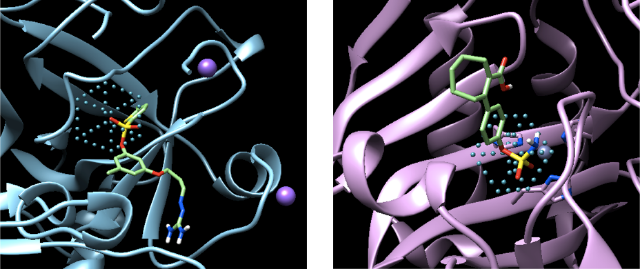 AtomNet® model learning to recognize sulfonyl groups – a structure often found in antibiotics.
AtomNet® model learning to recognize sulfonyl groups – a structure often found in antibiotics.
Critically, no human ever taught our AtomNet® technology the building blocks of organic chemistry. Our AtomNet® model discovered them itself by studying vast quantities of target and ligand data. The patterns it independently observed are so foundational that medicinal chemists often think about them, and they are studied in academic courses. Put simply, AtomNet® technology is teaching itself college chemistry.
AtomNet® technology can reproduce hundreds of historical experiments
Another way to test AtomNet® technology is to see if it could have predicted what happened in physical experiments done in the past. For this purpose, a group at the University of California, San Francisco developed a challenging benchmark, called DUD-E. This benchmark asks systems like our AtomNet® model to make over 1 million predictions, and compares the answers to the historical results. It is a hard and well-respected test, and our AtomNet® model achieves the best results of any structure-based algorithm we know of: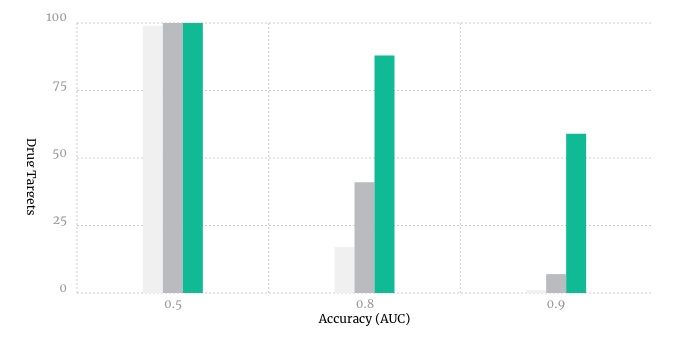 AtomNet® technology accuracy compared to previous technologies (DOCK and Autodock-smina) – on the DUD-E benchmark developed at UCSF.
AtomNet® technology accuracy compared to previous technologies (DOCK and Autodock-smina) – on the DUD-E benchmark developed at UCSF.
Put in real-world terms, AtomNet® technology's benchmark results suggest it could save something on the order of half of early stage drug screening experiments and greatly improve the success rate of many more.
AtomNet® technology has been predicting new potential treatments for two years
Our AtomNet® technology is not a hypothetical drug discovery platform. It has been in active use in real drug research for nearly two years. For many people, the gold standard for proving out a new discovery technique is, simply, using it to make important new discoveries. This also removes the possibility of teaching to the test, and has the upside that a prediction may lead to a life-saving medicine.
Our AtomNet® platform has already explored questions in cancer, neurological diseases, antivirals, antiparasitics, and antibiotics. Molecules predicted by AtomNet® technology have become the lead candidates in research programs, and produced positive results in animal studies. We are excited to continue this work and hope to keep sharing our discoveries as they develop.
 AtomNet® model predicting candidate treatments for Ebola – this prediction resulted in the lead molecule now awaiting animal trials.
AtomNet® model predicting candidate treatments for Ebola – this prediction resulted in the lead molecule now awaiting animal trials.
Caveats & Footnotes
- There are lots of downstream problems that this type of drug design does not address, mainly to do with how a drug is taken up in the body, breaks down over time, and causes side effects.
- Source: All That is Interesting – 30 GIFs That Explain The World Around Us
- Source: YouTube – Tipranavir Protease Inhibitor
- Source: Deep Learning and Applications in Neural Network