Our scientists have developed a set of chemical benchmarks to better understand exactly what our computational models are learning.
At Atomwise, AtomNet® structure-based models can quickly identify active compounds for protein targets from large chemical libraries for drug discovery projects. Understanding how models recognize features of molecular activity can shed light on, for example, important receptor-ligand interactions, biases toward particular functional groups, or target selectivity of compounds. However, it is still challenging to understand exactly what deep learning models are picking up during their training.
Atomwise scientists have developed a set of chemical benchmarks to assess what AtomNet® models are learning. Our graph convolutional architectures feature an attention mechanism, which can help scientists understand what molecular interactions the models focus on when learning a task. “The really big-picture idea of explainable AI projects like this one is to see if our model actually learns reasonable chemistry,” says Andreana Rosnik, a cheminformatics scientist at Atomwise.
To test if attention-based attribution provides quantitative information on how input features contribute to the network’s prediction, we first created a ground truth chemical benchmark set. Our team focused on recognizable molecular features that were simple and easy to compute, such as the number of hydrogen bonds in docked protein-ligand complexes. “Calculating hydrogen bonds with chemistry software tools is very straightforward so it stands to reason that a machine learning model should be able to do that easily too,” Rosnik says. “Most importantly, it's a question that we already know the answer to, and not an unknowable or complicated property. That makes it a great fit for a ground truth benchmark.”
There is another benefit to using the number of hydrogen bonds as a benchmark: they are usually correlated to some extent with how well a drug molecule fits into a binding site. “We put a lot of effort into making the models care about ligand poses,” Rosnik explains. “Examining hydrogen bonds can help us understand the intricacies of how our model differentiates between a good ligand-binding site fit and a bad one.”
Next, Rosnik curated a subset of publicly available data from the Protein Data Bank. The resulting data set contained several thousand target compounds and millions of associated protein-ligand poses and their bioactivity affinities that provided lots of examples of hydrogen bonds. Our team then compared the model’s predictions to the actual number of hydrogen bonds for particular protein-ligand poses. The predicted values were virtually identical to the true number of hydrogen bonds in the protein-ligand structures they assessed. Importantly, the AtomNet® model’s attention mechanism identified hydrogen bond donors and acceptors to a high degree of accuracy, validating the attention-based attribution approach used in our structure-based models.
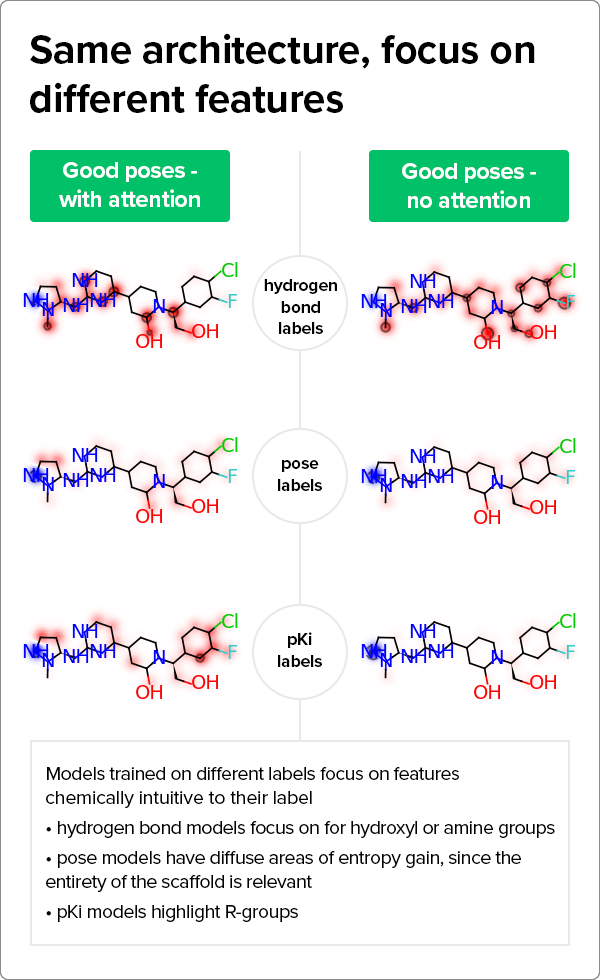
Strikingly, the models also learned important details about the components of the protein-ligand poses. “When we use the model to classify pose quality or predict pKi (binding affinity) values, we can see that it is paying attention to the things that we would expect it to for those labels as well,” Rosnik explains. “For example, when it is looking at a pose, it recognizes that there is more than a single atom involved. For pKis, it looks at the R-groups that confer the activity.”  There are several opportunities to expand on this project. For example, explainable AI can help predict structure-activity relationships at scale in drug discovery projects by quantifying how R-groups contribute to compound activity. “That will help our medicinal chemists build and evaluate hypotheses about large portions of chemical space, ultimately accelerating drug discovery timelines,” Rosnik says.
There are several opportunities to expand on this project. For example, explainable AI can help predict structure-activity relationships at scale in drug discovery projects by quantifying how R-groups contribute to compound activity. “That will help our medicinal chemists build and evaluate hypotheses about large portions of chemical space, ultimately accelerating drug discovery timelines,” Rosnik says.
Presentation: Dr. Andreana Rosnik was selected to present on this topic at the American Chemical Society 2022 Spring meeting. Take a deeper dive by viewing her presentation deck: https://blog.atomwise.com/synthetic-benchmarks-to-evaluate-attribution-for-structure-based-graph-convolutional-networks-in-drug-discovery
About Atomwise
Atomwise is a technology-enabled pharmaceutical company leveraging the power of AI to revolutionize small molecule drug discovery. The Atomwise team invented the use of deep learning for structure-based drug design; this technology underpins Atomwise’s best-in-class AI discovery engine, which is differentiated by its ability to find and optimize novel chemical matter.
Atomwise has extensively validated its discovery engine, delivering success in over 185 projects to-date including a wide-variety of protein types and numerous “hard-to-drug” targets. Atomwise is building a wholly-owned pipeline of small-molecule drug candidates, with three programs in lead-optimization and over 30 programs in discovery.
The company has raised over $174 million from leading venture capital firms to advance its mission to make better medicines, faster.
Learn more at atomwise.com, or connect on Twitter and LinkedIn.
