Our researchers are developing an automated pipeline that can more accurately identify promising compounds targeting multi-site proteins
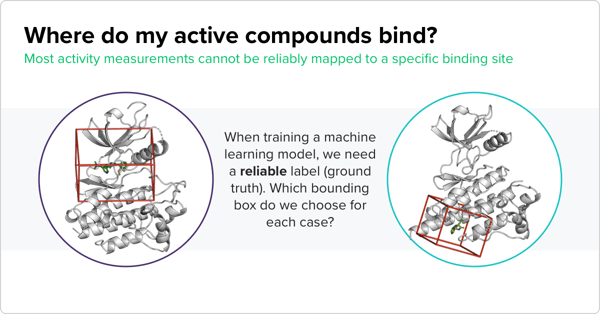
Applying deep learning to structure-based drug discovery provides a path for pharmaceutical companies to identify promising new compounds. These methods are likely to have a larger impact in research areas that have been challenging for pharma companies, such as studies focused on identifying compounds for target proteins with multiple binding sites. However, using these computational methods requires large quantities of well-annotated data regarding compounds and the protein sites they bind. Unfortunately, most compound activity measurements are not mapped directly to the exact binding sites on the protein. As a result, there is just not enough data annotated at the site level available in public databases such as the Protein Data Bank (PDB) or ChEMBL.
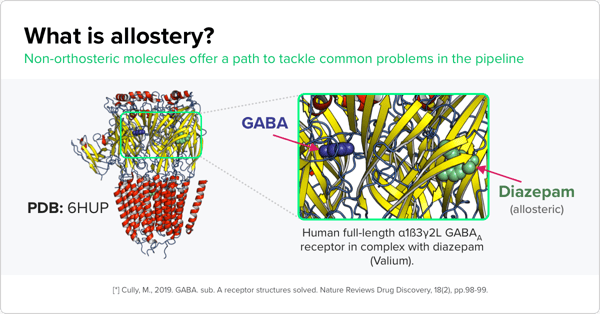
This makes it challenging to study allosteric sites, which are alternate positions on proteins outside of the active, or orthosteric, site that can modulate the protein’s function in some way. These are valuable sites for structure-based drug discovery initiatives because they could serve as promising drug targets.
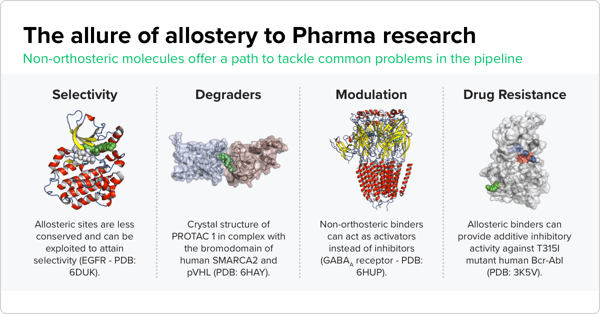
Pharmaceutical companies are interested in allosteric sites for several reasons. For instance, compounds that bind to allosteric sites can inhibit the activity of mutated proteins that are tied to disease. But the biggest driver for pharma is the likelihood of developing more selective drug compounds that pinpoint specific proteins. “Orthosteric sites are well conserved, while allosteric sites are less conserved,” Saulo de Oliveira, a cheminformatics scientist at Atomwise who is working on this project, says. “If you have a protein family with many proteins that share a similar, well-conserved binding site, for instance, the ATP binding site of protein kinases, one way to attain specificity — and make sure that your compounds are binding to one kinase versus another — is to target allosteric sites which are less conserved across the protein family.”
To address the challenge of targeting these sites, Atomwise is developing a fully automated binding site annotation pipeline that can identify the most likely binding sites for compounds with measured activity for known multi-site targets. To design the pipeline, Atomwise researchers had to address a key challenge — the need for properly annotated data to train their models. “Our tech is based on deep learning, which is very data hungry but also reliant on having accurate labels,” says Oliveira. Currently, compounds and proteins are matched using measurements of binding activity. This is a relatively simple task for cases where the majority of compounds being tested bind to a single protein site. But there are plenty of proteins where various compounds can bind to one of multiple possible sites. Using incorrect binding site information to train predictive models leads to poor model performance.
 For Oliveira, projects like this are his sweet spot. “Most of my interests have focused on the interface of biology and computer science,” he says. “I did a lot of structural bioinformatics so I have a unique skill set that allows me to process a lot of biological data, which is half of the equation.” To train the predictive models, he ensured that they had access to accurate data. As many as one in five compounds that lacked clear annotation were incorrectly assigned to a primary protein site, and the performance of the models was adversely affected.
For Oliveira, projects like this are his sweet spot. “Most of my interests have focused on the interface of biology and computer science,” he says. “I did a lot of structural bioinformatics so I have a unique skill set that allows me to process a lot of biological data, which is half of the equation.” To train the predictive models, he ensured that they had access to accurate data. As many as one in five compounds that lacked clear annotation were incorrectly assigned to a primary protein site, and the performance of the models was adversely affected.
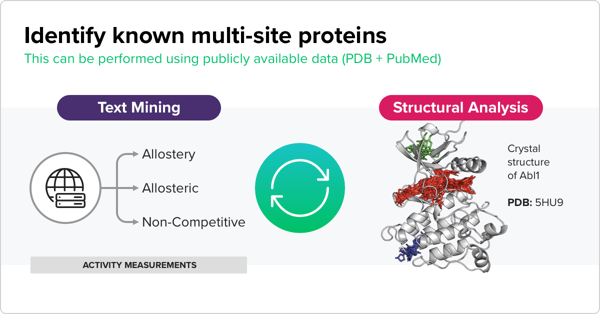
“If you make an incorrect guess at an early stage of the project, errors are going to propagate throughout the pipeline,” Oliveira explains. To avoid this issue, his team aggregated as much information on protein structure and binding sites as possible. This data would allow the models to make the best guesses regarding binding activity. His team took protein structure data from the PDB and activity measurement data gleaned from publicly available sources such as the ChEMBL database. They also pulled data on compound-protein complexes and considered protein homology, since an allosteric site on one protein is likely mirrored on homologs of that protein.
Combining all this data allowed Oliveira to train computational models that could make reasonable inferences about the binding sites. For example, he says, “if we have this compound that has a measured activity for a target but we don’t know if it's for site A or B, we look at all the compounds that bind to site A and those to site B. And we say ‘does this compound look like anything bound to either of those sites?’ and use that to make the decision.”
The data set included more than 500 multi-site proteins and more than half a million compounds. About 18% of compounds mapped to non-primary binding sites on proteins. The data was used to train and validate two kinds of deep learning models — virtual high-throughput screening and binding pose prediction. For both kinds of models, the more detailed annotations improved prediction performance for compounds binding to non-primary sites. Importantly, the models are significantly more accurate than existing methods such as molecular docking.
Learn More
Dr. Saulo de Oliveira was selected to present on this topic at the American Chemical Society Fall 2021 Virtual Meeting & Expo. Take a deeper dive by viewing his presentation deck: Automated Assignment of Active Compounds to Non-Primary Sites Helps Deep-learning Uncover Allosteric Modulators
About Atomwise
Atomwise is a technology-enabled pharmaceutical company leveraging the power of AI to revolutionize small molecule drug discovery. The Atomwise team invented the use of deep learning for structure-based drug design; this technology underpins Atomwise’s best-in-class AI discovery engine, which is differentiated by its ability to find and optimize novel chemical matter.
Atomwise has extensively validated its discovery engine, delivering success in over 185 projects to-date including a wide-variety of protein types and numerous “hard-to-drug” targets. Atomwise is building a wholly-owned pipeline of small-molecule drug candidates, with three programs in lead-optimization and over 30 programs in discovery.
The company has raised over $174 million from leading venture capital firms to advance its mission to make better medicines, faster.
Learn more at atomwise.com, or connect on Twitter and LinkedIn.
