The Atomwise approach to drug discovery is based on the strong belief that virtual screening enabled by sophisticated AI-based computational models will accelerate the development of new drug candidates, and eventually achieve our mission of creating better medicines that improve human health. Toward this pursuit our team has focused on one critical machine learning problem: the prediction of the strength of interaction between a compound and a protein based on the three-dimensional atomic structure of their bound complex.
We caught up with Venkatesh Mysore, principal cheminformatics scientist at Atomwise who straddles the machine learning and software engineering teams, to learn about his role in scaling up our AtomNet® platform architecture, and how solving the “Billion Trawler Problem” has been critical for advancing our drug discovery capabilities.
How did you first get interested in this challenge of scaling up our virtual screening capacity?
Since its inception, the collective intellectual thrust of Atomwise has been directed at the development of our AtomNet® technology, with the hope of bringing to life a platform for AI-based drug discovery that can accurately screen far more compounds on a computer than would ever be feasible in a wet lab. The realization of this vision became apparent when across several protein classes including targets that did not have an experimentally determined structure, we demonstrated to our partners and collaborators the ability to find assay-validated hits reliably.
Over the last two years, these virtual catalogues of compounds that could be synthesized on demand suddenly grew to the billions, with so many completely new compounds never tested against pharmaceutically relevant targets (or even synthesized before). Effectively, this meant that there were now thousands of haystacks, each with tens of needles — some way sharper than others. That led to the billion-dollar question: could we search these ultra-large catalogues to find the next blockbuster drug?
If we conquered millions of compounds with our AtomNet® technology, surely we must be able to screen billions of compounds. But that scale-up was a huge computational leap. We needed a shortcut, but it had to be a scientifically grounded shortcut.
So how did you find that shortcut?
Our team had an idea that a second “proxy” model that predicted the AtomNet® model scores rather than the experimental results could be a faster and more cost effective alternative to scoring every compound on the AtomNet® model. We developed that into an optimized workflow, fine-tuning each step of the protocol to maximize the ability to recover the truly top-scoring compounds that would have been found had the AtomNet® model been used to exhaustively score the library.
For a new project coming in, how does that work?
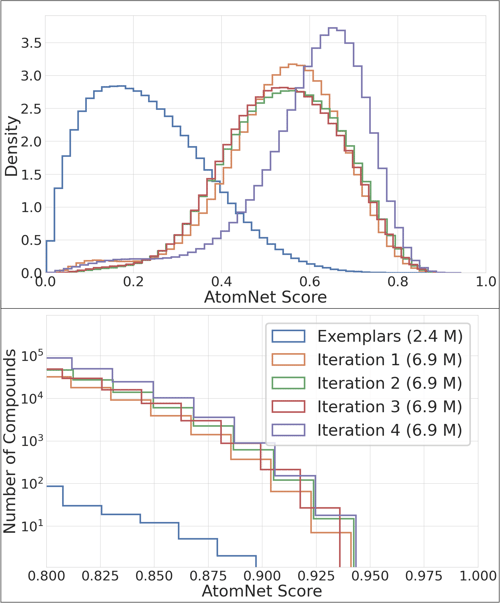
In the final ligand-based explorer, a small subset of carefully selected compounds representative of the entire library is run through the AtomNet® model every time we start working on a new target. Those compounds and their resulting AtomNet® model scores are then used to build a new machine learning model outside of the AtomNet® model architecture. We call it “the billion trawler answer,” and it aims to predict how new compounds would be scored by the AtomNet® model by comparing them to those that were already scored. The compounds predicted to do very well are then dispatched to the AtomNet® model, and their newly obtained scores are once again fed into the secondary proxy model. This process is repeated several times until the AtomNet® model has scored millions of compounds, with the faster and less expensive trawler model predicting the scores for the full catalog of billions. This iterative process, akin to active learning, greatly boosts the predictive power of the trawler model. With each iteration, the model becomes more and more accurate.
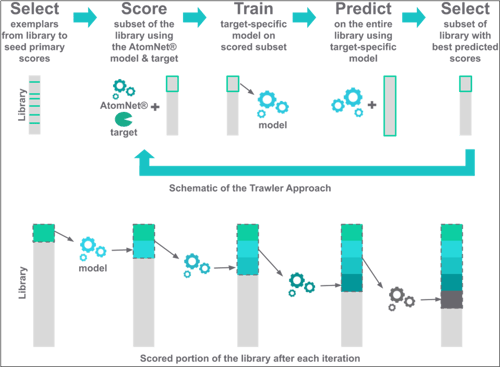
What was the development of this new approach like?
We had been noodling around with a version of the trawler model that ran reliably on million-compound catalogs for a while when, without warning, a new project came to Atomwise that required a billion-molecule screen. I literally had 10 days to code this. For those 10 days, I barely slept, implementing one fix after another to handle the memory and speed issues resulting from a thousand-fold increase in the search space. I remember anxiously watching over this nascent model trawling through billions of compounds for the first time. It was a mad rush, but it was definitely worth it.
Is that the model that Atomwise is using now?
No, after that sprint, our technical team worked to rebuild the workflow to make it more robust, stable, and scalable. The trawler originally employed random forests for the proxy model, but after evaluating gradient-boosted trees, graph convolutional networks, and fully connected neural networks, we chose GPU-based fully connected neural networks for production mode.
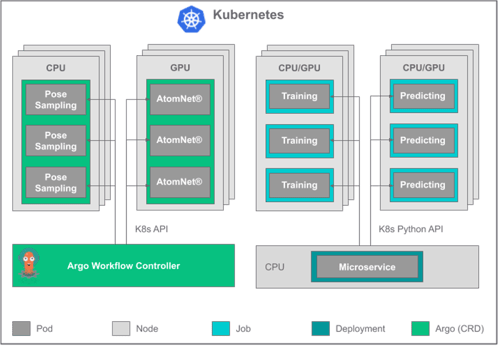
How has the trawler model improved screening capacity for customers?
Today, the trawler solution has been deployed for dozens of projects, sometimes screening over 16 billion compounds each time in only a few days. It has achieved up to a 500-fold increase in screening efficiency. It has also been incorporated into our routine screens of million-compound catalogues, with a conservative 10- to 20-fold speed-up in more than 100 projects. All told, this new technology has enabled our team to take up hundreds of new drug discovery projects, in addition to providing a practical way to mine the vast chemical universe for drugs of the future.
Learn More
Venkat Mysore, PhD, was selected to present a poster on this project and his work in scaling the AtomNet® platform at the American Chemical Society Fall 2020 Virtual Meeting & Expo. Take a deeper dive into his work by listening to the audio presentation and viewing the poster - Machine Learning Scales Virtual Screens to >10B Compound Libraries
Venkat Mysore, PhD also presented on this topic at the NIH Virtual Workshop on Ultra-Large Chemistry Databases on Dec. 1st. See his presentation deck - “Screening Billions of Compounds on the AtomNet Model: Approaches and Future Directions”
About Atomwise
Atomwise is revolutionizing how drugs are discovered with AI. We invented the use of deep learning for structure-based drug discovery, today developing a pipeline of small-molecule drug candidates advancing into preclinical studies. Our AtomNet® technology has been used to unlock more undruggable targets than any other AI drug discovery platform. We are tackling over 600 unique disease targets across 775 collaborations spanning more than 250 partners around the world. Our portfolio of joint ventures and partnerships with leading pharmaceutical, agrochemical, and emerging biotechnology companies represents a collective deal value approaching $7 billion. Atomwise has raised over $174 million from leading venture capital firms to advance our mission to make better medicines, faster.
