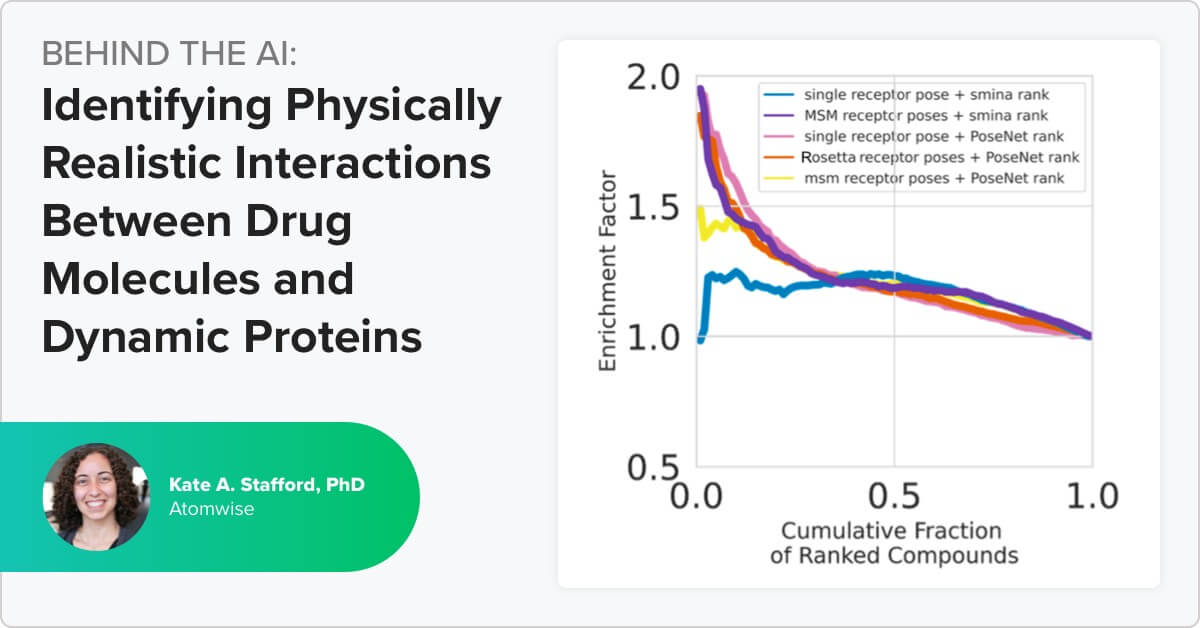
Our scientists developed AtomNet® PoseRanker to improve our technology’s ability to rank poses for protein-ligand interactions with potential drug compounds Identifying the highest-quality protein-ligand poses generated from structure-based virtual […]

Our scientists have developed a set of chemical benchmarks to better understand exactly what our computational models are learning. At Atomwise, AtomNet® structure-based models can quickly identify active compounds for protein targets from large […]
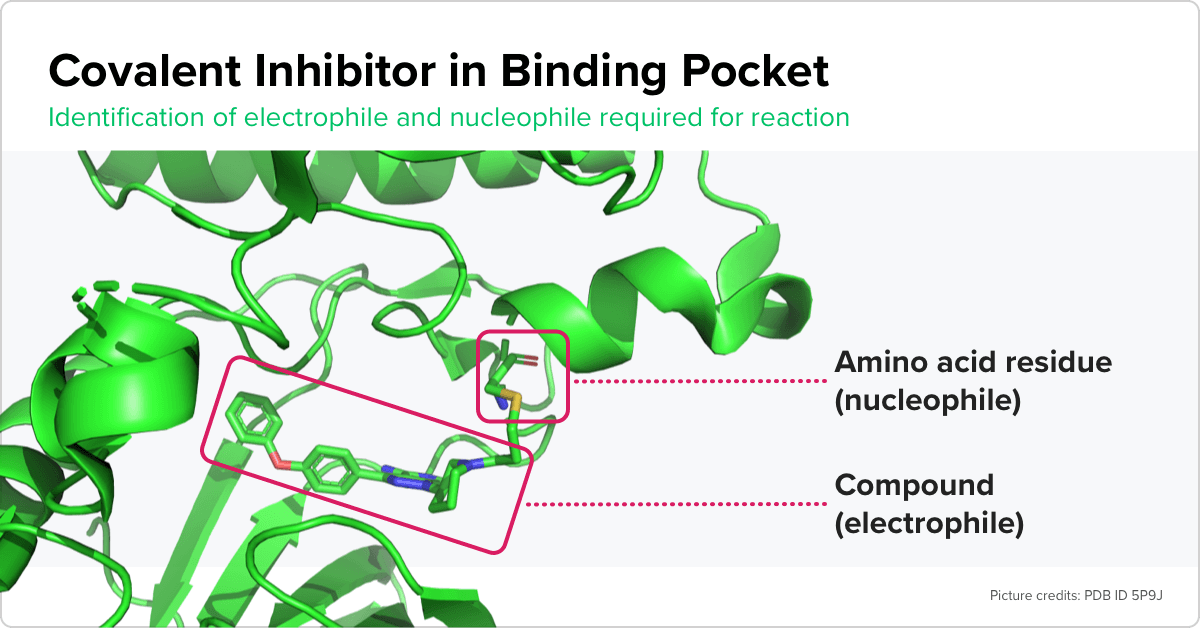
Atomwise scientists are adapting machine learning models to make predictions about a class of drug compounds that has been challenging to study.
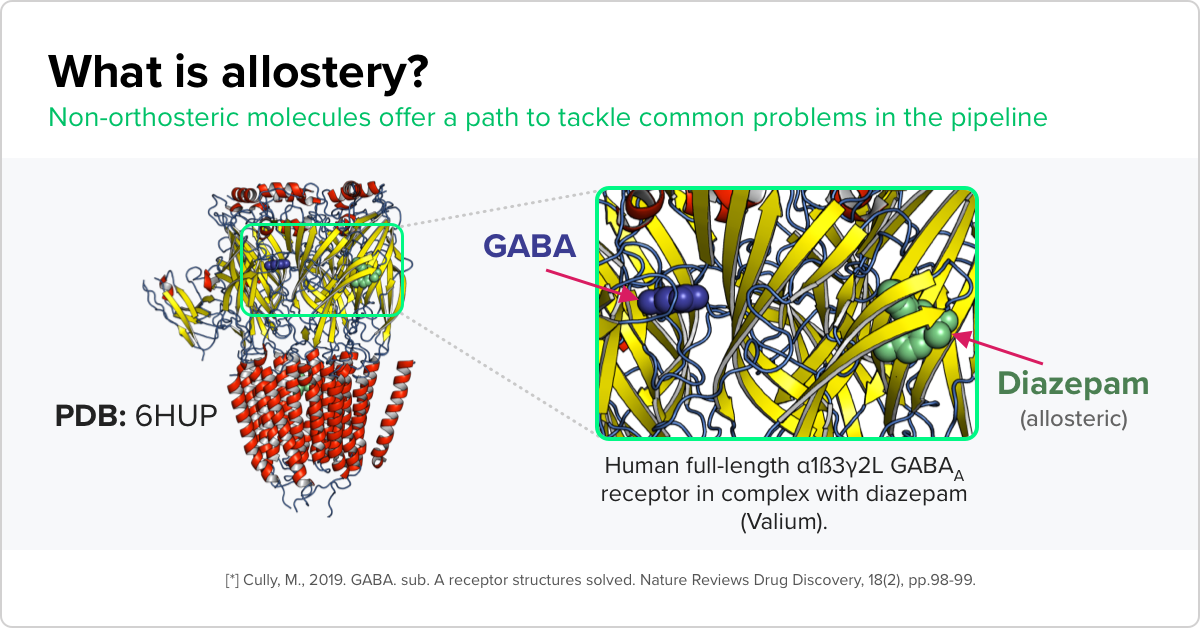
Our researchers are developing an automated pipeline that can more accurately identify promising compounds targeting multi-site proteins
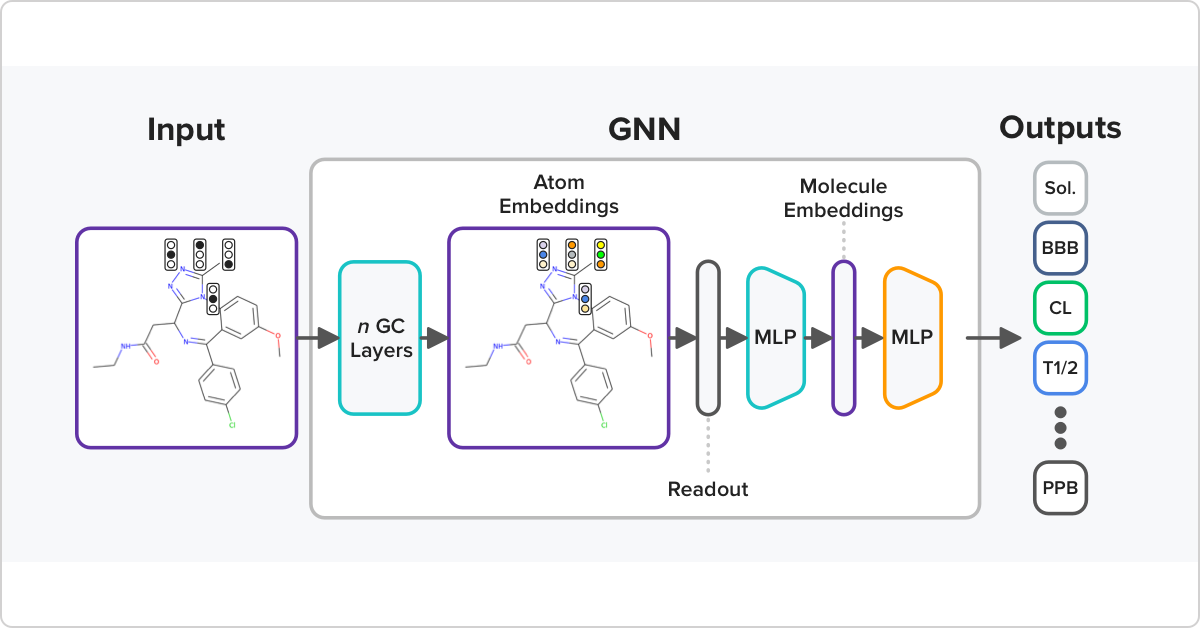
Atomwise scientists are improving predictive models for ADMET profiling by including a pre-training step that builds on diverse publicly available datasets
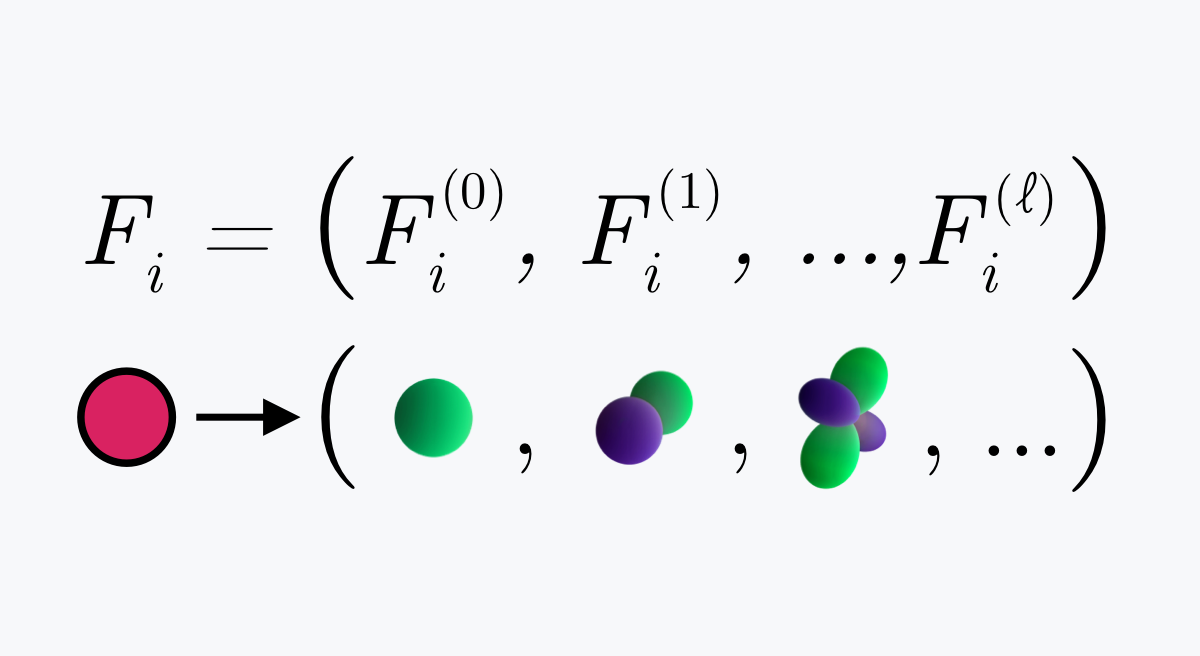
With the programmers on the Atomwise team, we have a steady stream of new ideas for how to improve the AtomNet® platform we created to virtually screen protein targets against billions of compounds to find promising drug candidates.
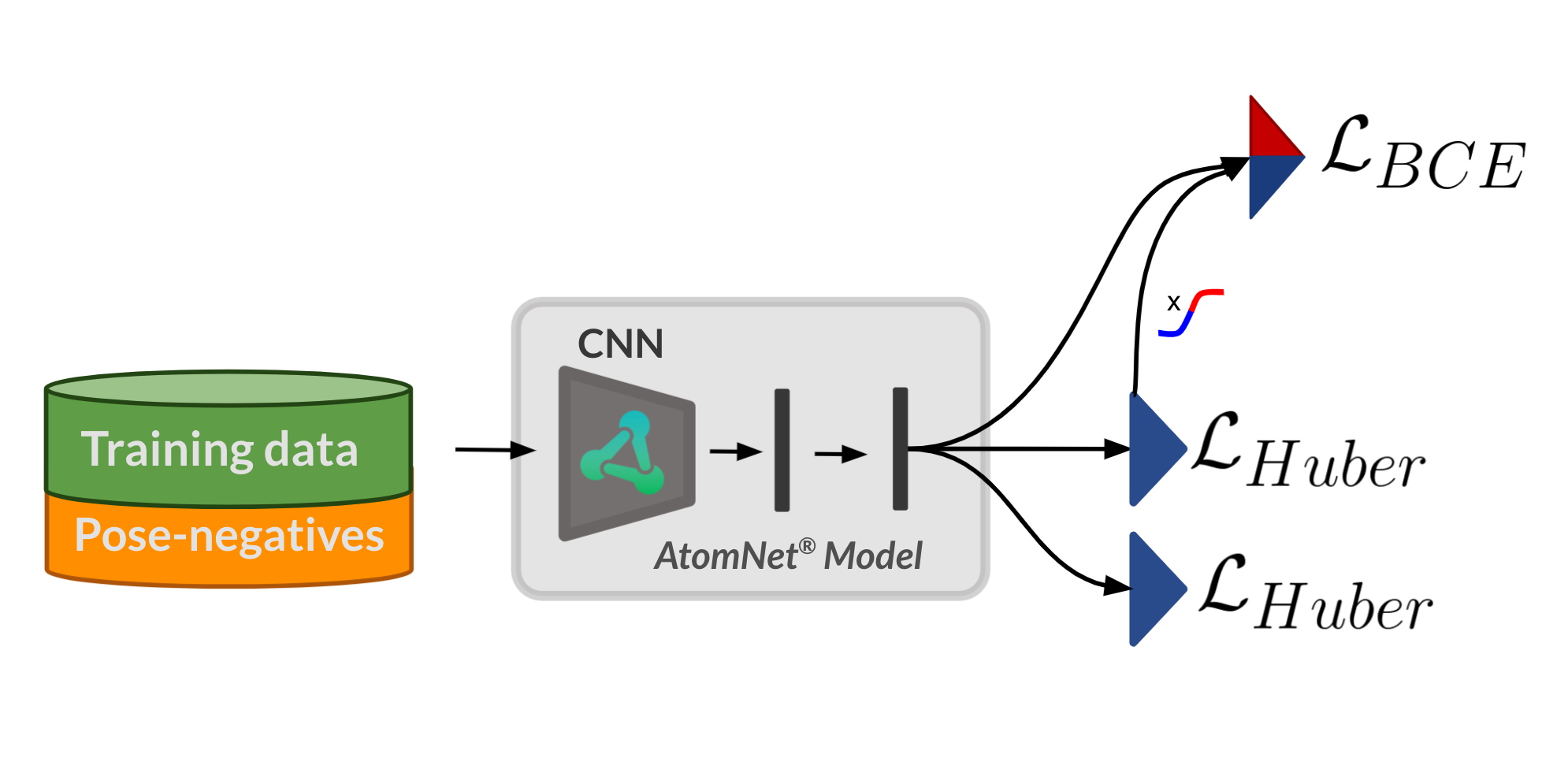
When you’re developing deep learning models, it’s not always obvious why a model performs the way it does. After all, the whole point of deep learning is to let the network teach itself from training data — and the way it assimilates that […]
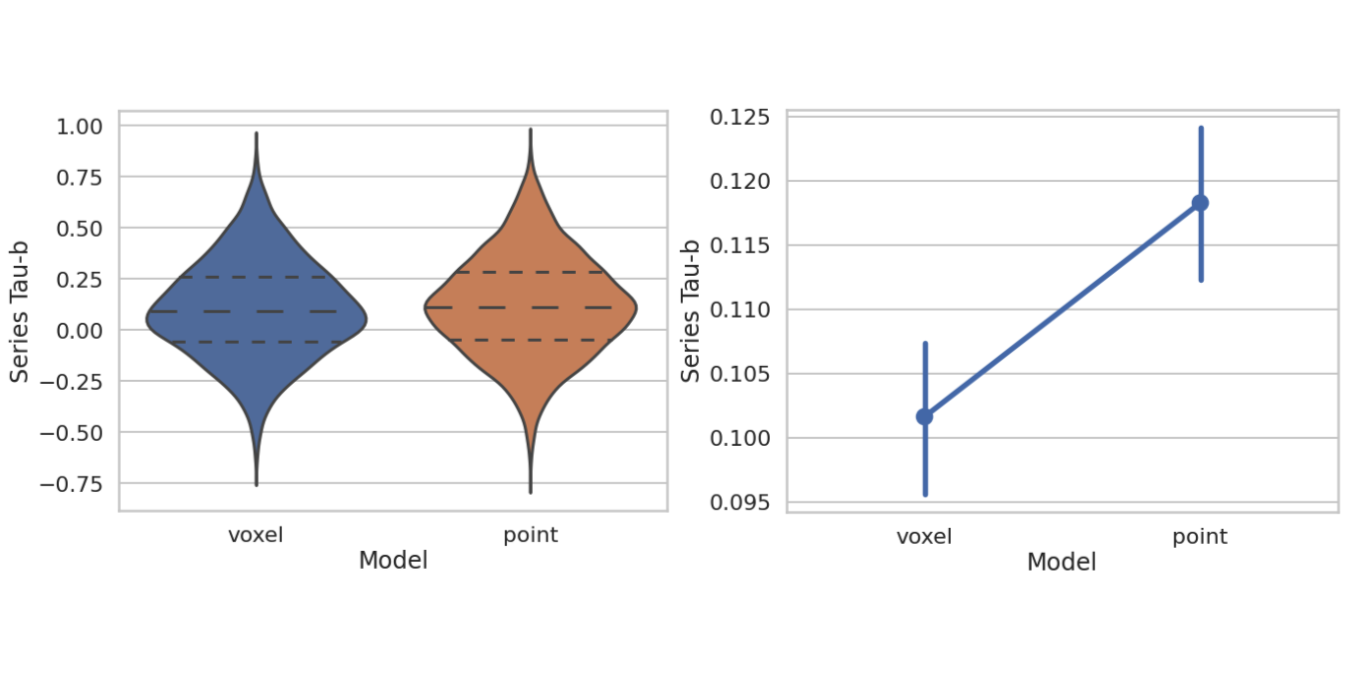
If you think of virtual drug screening as a video game, with players lining up models of compounds and protein targets to see if they fit together, then binding affinity might be how the game is scored. In drug discovery, we look for strong binding […]
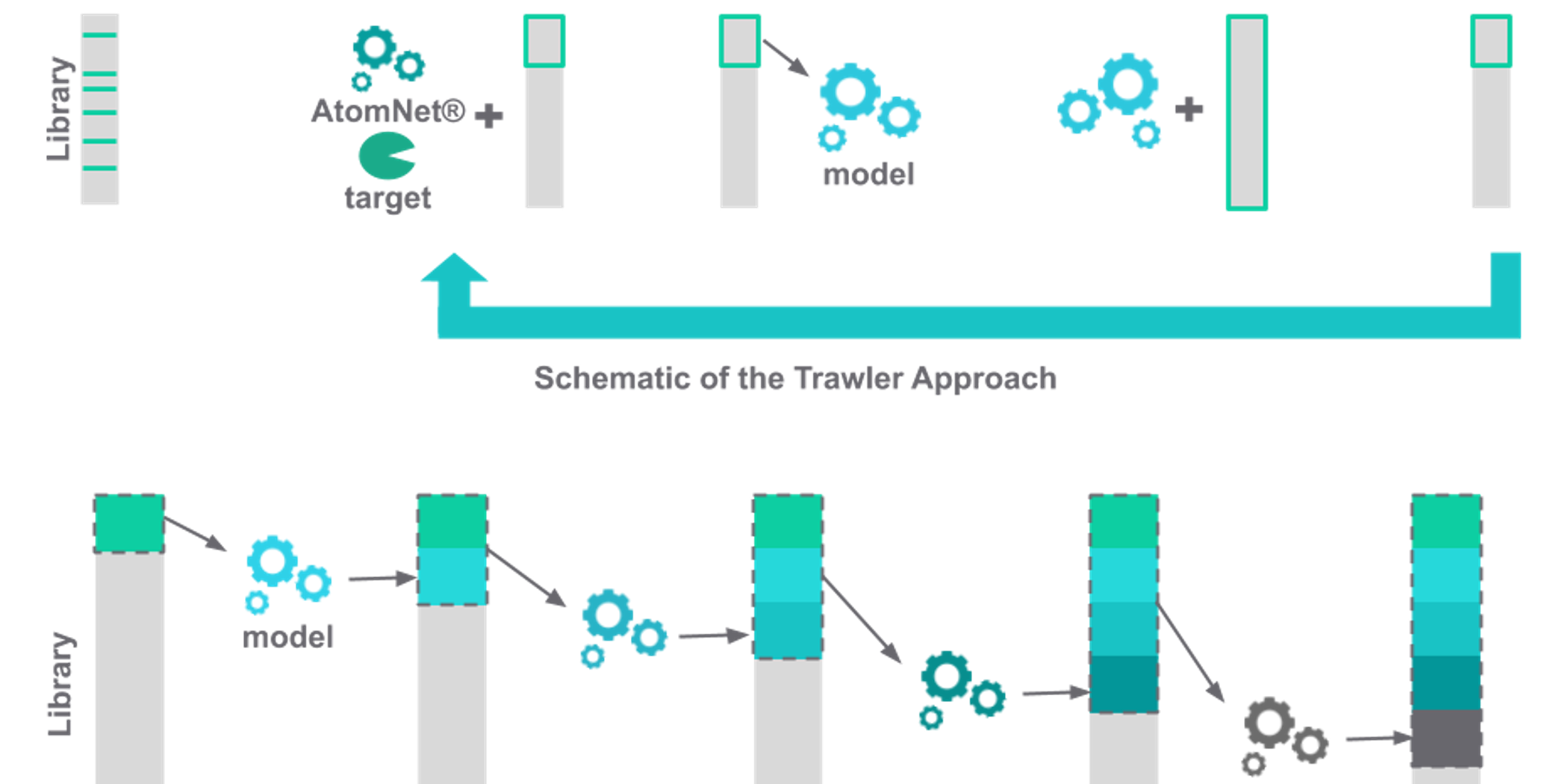
The Atomwise approach to drug discovery is based on the strong belief that virtual screening enabled by sophisticated AI-based computational models will accelerate the development of new drug candidates, and eventually achieve our mission of […]